APPRIS is a system [1] that deploys a range of computational methods to provide annotations of alternative splice isoforms and identify principal isoforms for multiple species. These annotations are based on the modules of the APPRIS Database [2].
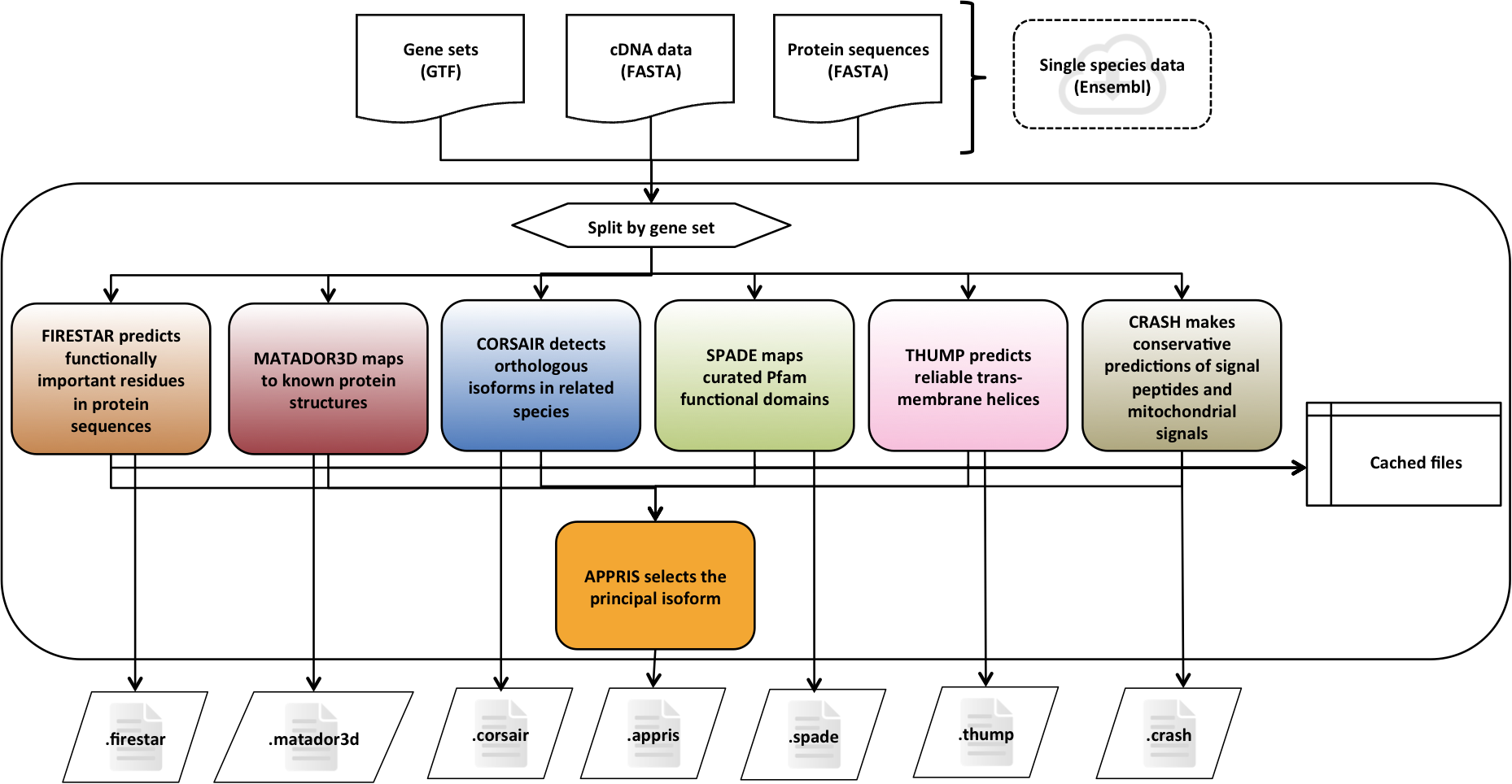
firestar predicts functionally important residues based on the fireDB database. The predictions are based on the local evaluation of alignments between the query sequence and the structures with functional information that are stored in FireDB. The reliability of predictions is assessed with SQUARE and the functional information is highlighted along with a reliability score.
Functional residues are highly conserved, even across large evolutionary distances. Since these residues are unlikely to have arisen by chance we can also use this to help determine the principal isoform.
Variants that have "lost" conserved functional residues are unlikely to be the principal isoform.
Protein structural information is analysed with Matador3D. In practice variant sequences from the same gene are mapped onto 3D structures by running BLAST against the PDB.
Protein structure is much more conserved than sequence and isoforms with large inserts or deletions relative to homologous crystal structures are also not likely to be principal.
Proteins are generally comprised of one or more functional regions commonly termed domains. Identifying the functional domains present in a variant can provide insights into the function and to determine the most likely principal isoform.
The presence of functional domains is analysed with Pfamscan.
Transcripts that have "lost" conserved functional domains are unlikely to be principal isoforms.
CORSAIR carries out BLAST searches against vertebrates to determine the most likely principal isoform. The method counts the number of species that align correctly and without gaps for each variant.
Transcripts that are conserved over greater evolutionary distances are more likely to be the main variant.
THUMP makes unanimous predictions of trans-membrane helices using three different methods: MEMSAT 3.0, Phobius, and PRODIV. A helix has to be predicted by all three methods to be considered reliable.
Transcripts that have "lost" trans-membrane helices are less likely to be the principal isoform.
The presence and location of signal peptides and cleavage sites in amino acid sequences are analysed with SignalP program. And TargetP predicts the sub-cellular location of eukaryotic proteins. CRASH uses a rule-based analysis of these two programs to select only reliable predictions.
The selection of principal isoforms coincides with the main isoform detected in proteomics experiments for almost 99.5% of comparable genes, showing that APPRIS principal isoforms are a clear improvement on choosing the longest isoform as the main protein isoform in the cell [3].
The data was generated by Spanish National Cancer Research Centre (CNIO) and Spanish Institute of Bioinformatics using a computational pipeline developed by Michael Tress and Jose Manuel Rodriguez.
APPRIS was developed within the GENCODE consortium [4] to address the challenge of annotating alternative protein isoforms of the Human genome, but it has been also annotated for another species.
If you have questions or comments, please write to:
All data is freely available to the public.
[1] APPRIS WebServer and WebServices. Rodriguez JM, Carro A, Valencia A, Tress ML. Nucleic Acids Res. 2015 May 18. doi: 10.1093/nar/gkv512
[2] APPRIS: annotation of principal and alternative splice isoforms. Rodriguez JM, Maietta P, Ezkurdia I, Pietrelli A, Wesselink JJ, Lopez G, Valencia A and Tress ML. Nucleic acids research 2013;41;Database issue;D110-7. doi: 10.1093/nar/gks1058
[3] Most highly expressed protein-coding genes have a single dominant isoform. Ezkurdia I, Rodriguez JM, Carrillo-de Santa Pau E, Vazquez J, Valencia A, and Tress ML. Proteome Res. 2015 Apr 3;14(4):1880-7. doi: 10.1021/pr501286b
[4] GENCODE: the reference annotation for the ENCODE Project. Harrow J., Frankish A., Gonzalez J.M., Tapanari E., Diekhans M., Kokocinski F., Aken B.L., Barrell D., Zadissa A., Searle S., et al. Genome Res. 2012;22:1775-1789. doi: 10.1101/gr.135350.111